Everything you need to know about the FDA 510(k) submission
What is an FDA 510(k) submission - and how do we complete one?
It's a question asked by many medical device companies in their earliest days. The FDA's 510(k) submission process is, in a nutshell, a clearance process which every company wanting to bring a medium-risk medical device to the American market must pass through.
Almost half of all medical devices used in the United States every day will have passed through the 510 (k) route. There are about 3000 510(k) submissions a year, but 30% of 2022 submissions weren’t even accepted for initial review.
It's a long, complex pro cess which can catch medical device quality and regulatory professionals out - so we've assembled everything you need to know about the FDA's 510(k) submission process right here.
Table of Contents What is an FDA 510(k) submission? The 510(k) submission and medical device classes What is a 510(k) predicate device? Who needs to submit a 510(k)? American medical device manufacturers Representatives of non-US manufacturers Specification developers Repackers/relabellers The three types of 510(k) submission The FDA 510(k) submission process Finding your predicate device Building a quality management system Device testing Submission Cover sheets Summary documents Statement documents SE documents Safety documents Digital/electrical documents Performance documents The wait Clearance! Top 8 submission mistakes 5 top tips FAQs Accelerate your FDA 510(k) submission

What is an FDA 510(k) submission?
The FDA's 510(k) submission process is, in short, a quality and compliance barrier designed to only let safe, effective medical devices onto the U.S. market, and into contact with American patients.
The focus of the 510(k) process is to prove something called 'substantial equivalence'. In other words, the aim of the game is to prove to the FDA that the medical device you want to bring to market is broadly similar to another device that's already on the market , known as a predicate device.
The logic of your 510(k) submission is clear: your device is like this predicate device, and here's how. And since this predicate device is already on the market and doing its job safely, your device must be safe and effective too!
At Qualio, we're big on making quality and compliance as simple as possible. With that in mind, here's a nice easy definition that gets at the heart of what an FDA 510(k) submission is:
510(k) A technical file given to the FDA proving that your medical device is like another already on the market
Your 510(k) submission should:
1. Demonstrate your medical device is ‘substantially equivalent’ to a predicate already on the market
2. House detailed technical, safety and performance device/test information to show your device is safe and effective
3. Prove you have a robust medical device quality and risk management system
Now that's cleared up, let's dig a little deeper.
The 510(k) submission and medical device classes
The FDA classifies all medical devices marketed in the United States into three risk categories: Class I, low risk, up to Class III, high risk.

The 510(k) submission process is generally applied to so-called Class II devices, with a medium risk profile.
There are some caveats to this: in some very rare cases, Class I and III devices might require a 510(k) submission. But as a general rule of thumb, only Class II devices require one.
A 510(k) allows you to bring your Class II device to market without clinical trials, by proving the substantial predicate equivalence we've already discussed.
Medium-risk medical device requiring a 510(k) submission are those which come into sustained and significant patient contact.
Example devices include:
- Blood pressure cuffs
- Insulin pumps
- Electric wheelchairs
- Pregnancy test kits
- Blood transfusion kits
- Contact lenses
- Surgical gloves
- Absorbable sutures
The first step of any 510(k) submission process is to be sure that it applies to you, and that your device isn't exempt from the 510(k) process .
Giving yourself a clear understanding of when to submit a 510(k) vs. a Premarket Approval (PMA) is also an important task to tick off early.
What is a 510(k) predicate device?
A predicate device is the existing marketed device which your 510(k) submission should prove is substantially equivalent to your device.
The predicate device should have:
1. The same intended use as your device
2. Similar technology to that involved in the function and operation of your device
3. The same level of safety and efficacy as your device
Your FDA 510(k) submission should prove all three of these criteria. If your device is definitely Class II and there’s really no substantial equivalent at all - which can happen if you have a really innovative medium-risk device - you’ll have to go down the de novo route .
510(k) completion can only happen with a suitable predicate at the core.
And of course, substantial equivalence is a slightly loose term which doesn’t mean ‘identical’. A purely identical medical device would offer no competitive difference or advantage to your business - so there's a tricky balance to strike here. Use your best judgment.
Who needs to submit a 510(k)?
There are 4 groups of actors who would, depending on specific business circumstances, be responsible for completion of an FDA 510(k) submission.
a. American medical device manufacturers
The most common 510(k) submitters.
Making a Class II device in the US and bringing it to market? The quality/ regulatory lead in your business would complete your 510(k) submission as part of your go-to-market activities.
b. Representatives of non-US manufacturers
The second most common 510(k) submitting group are the appointed representatives of medical device manufacturers outside the U.S.
FDA rules state that non-US manufacturers wanting to market their device in the United States require a representative to submit their 510(k) on their behalf.
c. Specification developers
Designed and developed a medical device but outsourcing production to a CMO?
The 510(k) submission is still your responsibility, not the CMO's.
d. Repackers/relabellers
It's not very common, but in certain cases repackers and relabellers in a medical device logistical supply chain could be responsible for a 510(k) submission.
This typically only happens as a kind of update 'special' submission (more on that below) in the event of a significant process change, such as if significant labelling changes are made (like adding a new use or warning to a manual), or if significant repacking changes are made in a way that could affect the safety and integrity of the device (like in a sterilization operation, for instance).
The three types of 510(k) submission
There are three types of 510(k) route:
%20submission%20types.png?width=1758&height=570&name=510(k)%20submission%20types.png "cover letter 510k")
It’s more than likely that you’ll fall into the traditional route, which is what we’ll be covering in detail here.
Don’t get too excited by the possibility of an ‘abbreviated’ route: this option actually takes around twice as long as the traditional process for the FDA to review and doesn’t require any less work on your part.
It’s only performed where you think it’s easier to prove that your device meets certain regulatory standards or guidance than it is to prove equivalence to a predicate.
The FDA 510(k) submission process
Now we've covered the basics of the FDA 510(k), let's take a look at the submission process itself.
a. Finding your predicate device
Don't overthink this first step. Just Google your medical device type and see what comes up!
Find an existing business and device and compare:
- Intended use
- Performance considerations: engineering, sterility, compatibility, software validation, etc.
This should give you a broad feel for your device category and how other potential predicate devices work.
Once you've done this, head to the FDA's 510(k) database . Do the same again: type in your device type and see what comes up.
Here, for instance, I can see 316 potential predicates for my pregnancy test kit:
%20database.png?width=601&height=397&name=FDA%20510(k)%20database.png "cover letter 510k")
Homing in on a single predicate device, I can see everything I need to start my 510(k):
%20predicate%20device.png?width=521&height=351&name=510(k)%20predicate%20device.png "cover letter 510k")
I’ve found a predicate device, and I can even see that that device itself has its own predicate device and was found to be ‘substantially equivalent’ (SESE) to another marketed pregnancy test.
Crucially, I can see the three-lettered product code (LCX) for this entire category of devices. So I can always go back and search this code if I want to check the entire group of substantially equivalent devices.
Remember: the wrong predicate device can scupper your submission and send you back to Square 1. So do some careful consideration here, always ensuring that the ‘substantial equivalence’ threshold is being met.
Read the official FDA guidance for determining substantial equivalence

Once you’ve found a predicate with provable substantial equivalence, use these key ingredients to learn as much as you can about your predicate device:
- 510(k) submission summary
- A sample (probably lots of samples…)
- Operating instructions
- Marketing literature
b. Building a quality management system
An effective medical device quality management system with documented, standardized processes and procedures is crucial for underpinning your 510(k) submission and proving your device's safety and integrity.
Before submitting your 510(k), you should focus on embedding compliance with FDA 21 CFR 820, as well as ISO 13485.
Your QMS should house clear SOPs for all of the following areas:
- Regulatory and compliance strategy
- General safety and performance requirements (GSPR)
- Management responsibility
- Resource management
- Risk management
- Performance evaluation
- Product realization
- Unique Device Identification (UDI)
- P ost-market surveillance
- Communication with competent authorities
- Incident reporting & field safety corrective action
- CAPA management
- Monitoring & measurement
Your QMS should also be supported by strong design , document and change control, with careful recording of your device lifecycle as the research, design and development stages unfold.
The more documentation you perform early, the easier your FDA 510(k) submission will be.

Learn more about the core requirements any medical device QMS needs for regulatory compliance
c. Device testing
Now we move onto the key 510(k) submission activity: performing your device tests to prove safety, efficacy and substantial equivalence.
The predicate device you chose in Step 1 will determine the tests you need to perform on your medical device. Your predicate is your benchmark and sets the acceptance criteria for your medical device testing.
Typical testing examples include:
- Mechanical/thermal/electrical
- Materials & make-up
- Wear/fatigue/durability
- Cleaning/sterilization
- Biocompatibility
- Electromagnetic compatibility
- Feature performance: normal and abnormal conditions
4 top testing tips 1. Use the product claims of your predicate device to set your test criteria 2. Use ISO 14971 as your benchmark 3. Look for historic recalls in your product category: use the faults as inspiration 4. Give yourself a digital mechanism for managing your design control activity
It's important that your business budgets wisely for this testing phase. Testing activity will form the bulk of your 510(k) expenses.
Get the right people and tools to maximize your in-house capability; typical third-party testing prices can quickly stack up:
- Third-party biocompatibility testing: $13,000
- Third-party sterilization testing: $15,000
- Third-party bench testing: $30,000
- Third-party electrical/EMC testing: $50,000
- Third-party implantation testing: $100,000+
If your device is an SaMD, you'll need to account for software validation activity too.
d. Submission
The FDA 510(k) submission itself is comprised of 20 key ingredients.
The FDA no longer requires a hard copy of the submission, and launched its Electronic Submission Template in September 2022 as part of a broad push towards digital submissions.
Following early success, in October 2022 the FDA announced its CDRH Customer Collaboration Portal would allow either an electronic copy (eCopy) or electronic Submission Template And Resource (eSTAR) 510(k) submission.
But now, after October 1, 2023, only the eSTAR is accepted. The eSTAR is a templated dynamic PDF you complete and submit electronically through the CDRH Portal (which you’ll need to sign up for). This more guided process is part of the FDA’s push to make 510(k)s faster and easier than ever before.
Let's dive into the key documents your 510(k) submission should include.
I. Cover sheets
The first two ingredients of your FDA 510(k) submission are your cover sheets:
- The FDA 3601 form (Medical Device User Fee Cover Sheet)
- The FDA 3514 form (CDRH Premarket Review Submission Cover Sheet)
The user fee cover sheet is your receipt for payment to enter the 510(k) process. The FDA website contains instructions for how to complete it.
The Premarket Review sheet is a basic summary of your submission, including company/device information, reason for submission, type of device, and so on.
Save $14,903 with a form Make less than $100m annually? Complete FDA Form 3602 (or 3602A for non-US firms) to register as a small business. Small businesses pay just $4,967 for 510(k) submissions in 2023, compared to the $19,870 standard fee. Businesses of all sizes also pay the annual establishment registration fee of $6,493 . Complete the Form 3602 every year to maintain small business status!
II. Summary documents
Next come your summary documents:
- Cover letter
- Statement of indications for use
- 510(k) summary OR statement
Your cover letter is a broad summary of your 510(k) submission.
Your indications of use statement MUST match the indications for use of your predicate device. It's made public after 30 days – so don’t reveal too much if your device is designed to fill a market niche!
For the final summary document, choose EITHER:
- Summary : combination of information that your SE claim is based on
- Statement : certification that you’ll provide safety/effectiveness information to any requester within 30 days
III. Statement documents
The 510(k) 'statement' documents are:
- Truthful & accurate statement
- Class III summary
- Financial certification/disclosure statement
- Declaration of conformity & summary reports
Not all of these documents may be relevant to you, depending on the circumstances of your submission.
The truthful and accurate statement is a standard, templated declaration as follows:
"I certify that, in my capacity as (the position held in company) of (company name), I believe to the best of my knowledge, that all data and information submitted in the premarket notification are truthful and accurate and that no material fact has been omitted. (Signature, date, time)."
As we’ve seen, most 510(k) submissions are for Class II devices - so the Class III summary is a simple declaration that you aren’t a Class III device. If yours is one of the rare Class III 510(k) submissions, you’ll need extra safety/effectiveness data here in line with your device’s higher risk profile.
The financial statements are only required if you conducted clinical trials for your medical device.
If you paid your investigators, complete the disclosure statement.
If you didn't, complete the financial certification.
And finally, the d eclaration of conformity & summary reports aren't required for the Traditional 510(k) route - they're for showing conformity with relevant standards in the Abbreviated route.
IV. SE documents
The SE documents, as you might expect, are for proving your device's substantial equivalence to its predicate.
We do this with three separate documents:
- Executive summary
- Device description
- Substantial equivalence comparison
The executive summary is a more detailed version of your 510(k) summary, elaborating on your completed testing activity.
Your device description should include your key technical information: Design History File, design outputs, drawings, specs, dimensions, and so on.
And your SE comparison is a side-by-side mapping of your device and its predicate, including the use, technology and performance of both devices.
V. Safety documents
The safety documents in your FDA 510(k) submission are:
- Proposed labeling
- Sterilization and shelf life
Your labeling proposal should include your device label, patient labeling, in-package inserts and operating instructions. Your Unique Device Identification (UDI) code isn't mandatory at this stage, but you should include a sample barcode of where it will go.
If your device isn't sterilized or with a shelf life, you can skip the next section. If it is, include your test data for these areas here.
And for the biocompatibility section, include your test protocols and reports for any areas in patient contact (direct or indirect).
VI. Digital/electrical documents
If your device has a digital or electronic component, you'll need to include these next sections, Sections 16 and 17, in your 510(k) submission. If not, don't ignore them - briefly explain why they aren't relevant for you.
There are two documents to consider here:
- Electromagnetic compatibility/ electrical safety
For the software documentation, establish your device's Level of Concern (LOC) and add your test data accordingly.
Helpful software reference documents Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices IEC 62304
Your EMC document should prove your device won’t interfere with, or be jeopardized by, other nearby devices.
The electrical safety document should prove that the electrical components in patient contact aren’t a threat to patient safety.
Helpful EMC/EC reference documents IEC 60601-1 IEC 60601-2
It’s important to note that your device can still be a hardware device and include a software element. If your device falls here, you still need to account for all software operation in your 510(k) submission, even if you aren’t an ‘SaMD’ or 'SiMD' by definition.
Firmware-related software will still need documentation; your device's Level of Concern will determine what level of documentation is needed.
%20device%20software%20hardware.png?width=606&height=522&name=510(k)%20device%20software%20hardware.png "cover letter 510k")
VII. Performance documents
Finally, we arrive at your device's performance documents. These are arranged in 3 sections:
Only the first is mandatory; the other two may not apply to your operation.
All completed tests should have protocol descriptions, summaries, methodologies and clear results.
Read FDA guidance on bench testing
e. The wait
You've assembled the sections of your 510(k) submission.
You've gone over the FDA's Refuse to Accept policy to make sure you're meeting all of its 56 requirements and won't receive a refuse-to-accept letter.
Now it's time to press the button and wait.
How long does an FDA 510(k) submission review take?
The FDA's official timeline is as follows:
%20timeline.png?width=222&height=407&name=FDA%20510(k)%20timeline.png "cover letter 510k")
Despite the ‘ideal’ 100-day timeframe, the average approval time after submission is 175 days .
This is skewed by some excessively long device approvals however, with the median approval time standing at 85 days .
The average number of days it takes to clear a device via 510(k) also varies according to the device category.
Anesthesiology devices have the longest average length to approval, averaging 245 days.
Toxicology devices are the shortest on average, at just 163 days. In short, your business is looking at a 3-6-month waiting time after submission.
A medical device is never 'approved' by the FDA after a 510(k) submission.
Instead, y ou’ll receive a letter (no certificate) confirming your device is cleared for marketization in the United States.
You can now market your device, but must still comply with other requirements like:
- Registration and listing (21 CFR Part 807)
- Labeling (21 CFR Part 801)
- Reporting (21 CFR 803)
- GMP requirements (21 CFR Part 820)
Well done! Pour yourself a drink.
Top 8 510(k) submission mistakes
As we've already seen, a full 30% of 510(k) submissions in 2022 weren’t even accepted for initial review. A right-first-time 510(k) approach gets you to market faster, eliminating wasted time and effort while generating revenue more quickly.
To help you, we've assembled the 8 most common mistakes made by medical device manufacturers in their FDA 510(k) submissions.
1. Incorrect templates/document versions
2. Choosing an unsuitable predicate
3. Inconsistent information in areas where information is repeated in the submission
4. Skipping non-applicable sections, instead of documenting why they aren’t applicable
5. Incomplete test information (summaries without protocols/reports)
6. Wasting money by not applying for small business status
7. Immature quality/risk management
8. Generally disorganized submission documents (no ToC, lax sectioning/numbering, etc.)
As an electronic quality management software provider, Qualio works with international medical device organizations every day. Our team is full of experts with decades of collective quality and regulatory experience.
To make your FDA 510(k) submission process even smoother, here are 5 top tips from the team.
1. Try and use a fairly recent device as a predicate. Devices older than a decade are frowned upon!
2. Ensure everything is covered - nearly two-thirds of submissions are slowed by Additional Information (AI) requests
3. The more design control, risk and quality documentation you can organically build as you develop your device, the faster and easier your 510(k) assembly will be
4. Substantial equivalence is everything! 15% of submissions fail because of the wrong predicate
5. Familiarize yourself with the eSTAR now, so you’re prepared for the October 2023 transition date
We've hosted a number of webinars about the 510(k) submission process, and welcomed hundreds of international attendees through our virtual doors.
Frequently asked questions (FAQs) are a valuable way to pinpoint areas of uncertainty and confusion - so we made a note and answered them!
"Can a non-US manufacturer submit a 510(k), or does it need to be done by a US representative?"
Yes, a foreign manufacturer can submit their 510(k) application directly to the FDA. For convenience you could get the help of a US-based contact, but it isn't mandatory.
"What exactly is a 'reserved' Class I device?"
Reserved Class I devices are those 'special case' devices that the FDA believes meet the reserved criteria in section 206 of the Modernization Act and, therefore, would require a 510(k) under section 510(l). View a list of reserved devices here .
"Can you discuss the differences in the process when submitting a 510(k) to CBER as opposed to CDRH?"
Combination products are typically marketed under a marketing authorization type associated with whatever constituent part provides the primary mode of action (PMOA).
Combination products with a drug PMOA need a new drug application (NDA) or abbreviated new drug application (ANDA).
Those with a biologic PMOA need a biologic license application (BLA).
And those with a device PMOA need a PMA, de novo or 510(k) submission, depending on risk profile.
A single marketing application is generally sufficient for a combination product. In some cases, however, a sponsor may wish to submit separate marketing applications for different constituent parts of a combination product, and the FDA may consider this permissible.
"How can you see the labeling of a predicate device?"
The best way would to try to locate an example on the market. Labels for such devices are also commonly placed in the 'about' section of the manufacturers's app or website.
"If my Class II device is exempted from 510(k), what documents and/or reports are required to market it in the US? Do we need to inform the FDA before marketing?"
All medical devices, even if exempt, are required to follow the Quality System Regulation (QSR) mapped out in 21 CFR 820 . In some cases, exempt devices may be exempt from Good Manufacturing Practice requirements under the QSR.
As for the specific documents/reports and the level of detail needed, this can vary based on your device and its intended use and claims.
"Are there any harmonized standards given like in the EU MDR?"
View the list of Recognized Consensus Standards on the FDA website here .
"I'm not sure how to determine if the predicate device we've found is appropriate for submission. What do I do?"
The FDA has a handy decision-making guidance document for this. View it here .
"Can we use a predicate device not marketed in the US?"
No, your predicate must be a US-marketed product.
"Can we use one of our own devices as a predicate?"
Yes you can, and lots of companies do! Just make sure it's substantially equivalent.
Accelerate your FDA 510(k) submission
Want a faster, more controlled 510(k) process?
Qualio provides electronic quality management software to help medical device manufacturers digitize, automate and strengthen their quality management activities.
Electronic documentation, controls, workflows and records simplify all aspects of your FDA 510(k) submission process and get you to market faster.

Alex has worked in the quality and compliance space for 5 years, producing a range of industry content to help Qualio blog visitors understand the complex and highly regulated environments of modern life science. Since graduating with a master's degree from the University of Cambridge, Alex has produced training courses, webinars, whitepapers, blog posts, e-books and more on a range of life science quality topics, from GxP to ISO 13485. Alex is passionate about the transformational power of a culture of quality and writes extensively about digital quality management, life science company growth and easing compliance burden.
Related Articles
4 regulations that apply to medical device cro selection, when to submit a 510(k) vs. a premarket approval, 510(k) exempt medical devices: how to tell if you need to submit.
DOWNLOAD COMPLETE 510(K) SUBMISSION GUIDE
Get cleared first time with our comprehensive submission roadmap guide

More than a Quality Management System: Tools for the entire MedTech Lifecycle.
Featured Capabilities:

Experience the #1 QMS software for medical device companies first-hand. Click through an interactive demo.
Accelerate development with integrated design control and risk software.

Schedule a custom demo of Greenlight Guru Product now.
Data collection and management designed for MedTech clinical trials.

Get a personalized demo of Greenlight Guru Clinical today.
- By Initiative
- Migrating From Paper
- Managing and Assessing Risk
- Preparing for Regulatory Submissions
- Becoming Audit Ready
- Managing Postmarket Quality
- Small Business
- By Function
- Product / R&D
- ROI Calculator
- Customer Success
- Case Studies
- Checklists & Templates
- eBooks & Guides
- Content Hub
- Live & Virtual Events
- True Quality Roadshow
- Quality Pricing
- Clinical Pricing

Preparing Your Abbreviated 510(k) Submission in 7 Easy Steps
%20Submission%20in%207%20Easy%20Steps.png "cover letter 510k")
The Abbreviated 510(k) pathway is one more tool to add to your tool belt for bringing a new medical device to market.
This pathway is still not widely-used - only around 2% of clearances have taken this route in recent times. However, it just might be the right path for you, and the more you know, the better you can choose a pathway to market that will be to your best strategic advantage.
What is it? An Abbreviated 510(k) is where you show substantial equivalence to a recognized standard, special control or guidance using a declaration of conformity. In other words, it’s a paper-based comparison , rather than a comparison with a specific device.
According to the FDA guidance , device manufacturers may choose to submit an Abbreviated 510(k) when the submission relies on one or more:
- FDA guidance document(s);
- Demonstration of compliance with special controls for the device type, either in a device-specific classification regulation or a special controls guidance document; and/or
- Voluntary consensus standard(s).
If you’ve determined that an Abbreviated 510(k) is the right pathway for your device, what are the next steps?
FREE CHECKLIST: Want to make sure you’ve included all necessary sections in your Abbreviated 510(k) submission to FDA? Click here for a free downloadable checklist that can help.
#1. Prepare “abbreviated 510(k)” cover sheet
A Medical Device User Fee Cover Sheet is required with your application. This answers a series of questions about the type of application being submitted to FDA and includes basic information about your company. The FDA recommends that even if your device has a fee exception, you should still include a cover sheet that explains what your exception is.
You’ll also need a 510(k) cover letter, which should summarize the type of 510(k) submission you are making. Typically, you will write the cover letter last, so as to provide a good, high-level summary. Your cover letter should include the following:
Type of 510(k) submission, Abbreviated or Traditional
Your device type in plain terms, i.e., by its common name
510(k) submitter
At least one contact person, by name, title, and phone number
Your preference for continued confidentiality (21 CFR 807.95)
Your recommended classification regulation
Class (i.e., whether it is unclassified or a class I, II, or III device)
Review panel
The FDA product code
Any FDA document numbers associated with prior formal correspondence with FDA (e.g., IDE, pre-IDE, 510(k), PMA, request for designation (RFD)) related to your device.
#2. Prepare description, intended use and indications for use
The recommended next step is an indication for use statement. You can use FDA Form 3881, which is used to identify and describe indications for use for a medical device. An important note here is that you need to be consistent. Your indications for use statement must be exactly the same as your indications for use listed throughout the rest of your submission.
You should also prepare a device description, including its intended use. Your intended use should match what you claim your device is for in labeling. It’s a balancing act as intended use also affects the classification of your device. A scalpel becomes Class III if its intended use is on the eyeball…
It’s important not to mix this up with indications for use, which are the reasons or situations for using the device. These must be clear in your submission.
#3. Prepare an abbreviated 510(k) summary or statement
A summary or statement is required for all types of 510(k) submission, including the abbreviated 510(k). You can choose either the summary or the statement, and can elect to change your choice right up UNTIL the substantial equivalence determination is made.
510(k) summary
This information should summarize everything for which you are basing your claim of substantial equivalence. In other words, it provides specific examples that you are citing as evidence. FDA places a summary online 30 days following the determination of substantial equivalence.
For submissions that rely on FDA guidance documents or are subject to special controls, you need a summary report that describes how those were used to demonstrate substantial equivalence.
You may also rely on voluntary consensus standards, for which you should consult the FDA’s guidance on the appropriate use of voluntary consensus standards.
510(k) statement
The 510(k) statement is really a promise that you will provide safety and effectiveness information to support an FDA finding of substantial equivalence to ANY person within 30 days of a written request.
If you elect to take this option, literally anyone can request a copy of your abbreviated 510(k) submission (with confidential information and trade secrets deleted). So it may be something of a strategic choice.
#4. Include a truthful and accurate statement in your abbreviated 510(k) submission
The “truthful and accurate statement” can be as simple as this:
I certify that, in my capacity as (the position held in the company) of
(company name), I believe to the best of my knowledge, that all data
and information submitted in the premarket notification are truthful and
accurate and that no material fact has been omitted.
_____________________________
(Signature)
#5. Prepare proposed device labeling in your abbreviated 510(k) submission
Labeling is also a required part of your abbreviated 510(k) submission, for which FDA says:
The 510(k) must include proposed labeling in sufficient detail to satisfy the requirements of 21 CFR 807.87(e). If the device is an in vitro diagnostic device, the labeling must satisfy the requirements of 21 CFR 809.10. Generally, the term “labeling” includes the device label, instructions for use, and any patient labeling. See the FDA guidances “Device Labeling Guidance #G91-1,”25 “Labeling – Regulatory Requirements for Medical Devices,”26 “Guidance on Medical Device Patient Labeling,”27 and device-specific guidance, where available, for more information about labeling your device.
(Note: that many parts of the abbreviated submission are exactly the same as the traditional 510(k) submission and can be found in this FDA guidance document ).
It’s important to know that FDA considers the term “labeling” to cover many things, from your packet inserts to your marketing. You can find a summary of these labeling types here .
#6. Add a specifications section to your abbreviated 510(k) submission
Your specifications section should have a narrative description and a physical description of your device. This includes performance specifications and a brief description of device design requirements. FDA recommends that you identify all models and accessories. They also state:
If diagrams, dimensions, tolerances, and/or schematics are useful to fully describe and characterize the device, we recommend that you include them for each device and accessory included in the 510(k) submission. We also recommend that you provide a list of all tissue-contacting components and their respective materials.
#7. Add a substantial equivalence comparison section
One of the recent changes to the FDA criteria for the abbreviated 510(k) is to allow companies to establish substantial equivalence through performance criteria. This relies on established performance requirements and associated test methods.
Essentially, in this section, you need to prove that your device meets the appropriate, established criteria and cite your sources. To go a step further, you might also talk about why something is not relevant to your case. Sometimes, saying why something is not relevant can save you from a back and forth discussion with the FDA.
Gain clearance for your abbreviated 510(k)
As with the traditional premarket notifications, an abbreviated 510(k) does require quite a bit of work. You need to have exhaustive evidence to back your claims and comprehensive documentation. You’ll gather this information from multiple areas of your QMS, which should be kept updated at all times.
For medical device companies, the strength of their QMS can make or break their FDA clearance hopes. Not all quality management systems are built the same - many are simply not purpose-built for what medical device companies need and can be cumbersome to set up, manage, and maintain.
The best QMS solution will be purpose-built for medical devices, making these tasks simple and easy to execute. Greenlight Guru is the QMS software built by medical device professionals for medical device professionals.
Your journey to getting your device cleared via the abbreviated 510(k) pathway can be made effortless with the implementation of our QMS software. Get your free personalized demo now →
Looking for a design control solution to help you bring safer medical devices to market faster with less risk? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software
Jesseca Lyons
Jesseca Lyons is a Senior Medical Device Guru at Greenlight Guru and a Mechanical Engineer by trade who loves working with cross functional teams, including both engineering and non-engineering disciplines. She’s spent most of her career gathering and defining requirements for new product design and development in the...
Related Posts
What is the estar pilot program and how will it improve fda's 510(k) review process, 510(k) tips and answers to frequently asked questions for medical device companies, common mistakes that can tank your fda 510(k) submission, subscribe to our blog.
Join 200,000+ other medical device professionals outperforming their peers.

Get your free checklist
Abbreviated 510(k) submission checklist.
%20Submission%20Checklist-1.png?width=250&name=Abbreviated%20510(k)%20Submission%20Checklist-1.png "cover letter 510k")
- Checklists/Templates
- Request a Demo
- Content Title Description
510k Electronic Submission Guidance for FDA 510k Submissions
This is an overview of the updated 510k electronic submission guidance document that the FDA released on October 2, 2023.
What’s included in the 510k electronic submission guidance?
As with any FDA guidance, there is an introduction and background regarding the reason for the updated guidance document (i.e., eSTAR guidance). At the very beginning of the document (i.e., page 3) the reference to the RTA Guidance was deleted, because there is no longer an RTA screening process with the implementation of the FDA eSTAR templates. The updated guidance explains on page 6 that “The CDRH Portal will automatically verify that the eSTAR is complete, and therefore we do not expect to receive incomplete 510(k) eSTARs.” In the scope section, the FDA specifies that this document is specific to 510k submissions using the eSTAR template. The document also explains that CBER conducted a pilot with the eSTAR template in June 2022 and now the FDA eSTAR template must be used in conjunction with the CDRH Portal for submission of a 510k to CBER. The FDA has plans to release a similar De Novo submission guidance for using the eSTAR template, but this has not happened in the year since the FDA announced the intention to do so. In the “Significant Terminology” section of the guidance (i.e., Section IV), the FDA provides definitions for each of the different types of submissions: eCopy, eSubmitter, etc. In the “Current Electronic Submission Template Structure, Format, and Use” section of the guidance (i.e., Section V), the FDA modified the term used for the company that is applying for 510k clearance from “Submitter” to “Applicant,” because sometimes a regulatory consultant or 3rd party reviewer is submitting the 510k on behalf of the applicant. On page 12 of the updated guidance, the FDA added “Withdrawal requests” to the list of 510k submissions/information that is exempt from the 510k electronic submission requirements. In the next to last section of the electronic submission guidance, the FDA provides a table outlining all of the sections of the new eSTAR template. The table is reproduced later in this article. If you are interested in a tutorial on completing each section outlined in the table, we recommend purchasing Medical Device Academy’s 510(k) Course . The last section of the eSTAR guidance indicates the timing for compliance with the updated guidance (i.e., October 1, 2023).

What is the deadline for compliance with the guidance?
The deadline has now passed. The new eSTAR template must be used for all 510k and De Novo submissions as of October 1, 2023. You must upload the new FDA eSTAR submissions using the CDRH Portal. You will need to request an account using a registration hyperlink .
What’s missing from this 510k submission guidance?
The updated 510k electronic submission guidance does not provide information regarding the receipt date for electronic submissions made through the new customer collaboration portal (CCP) created by CDRH. The image below is a screen capture of the current CCP upload webpage. It includes the following statement, “Send your submission before 16:00 ET on a business day for us to process it the same day.” This statement was added sometime in August or September, but the FDA has not released a detailed explanation. This statement makes it clear that the FDA is not promising to process a submission the “same day” if the submission is received after 4:00 p.m. ET. However, “processed” does not have the same meaning as “receipt date.”
Another element missing from this updated guidance is a reference to human factors documentation. For any devices that have a user interface that is different from the predicate device, and for software devices, the FDA requires documentation of your human factors process to make sure that differences in the user interface do not result in new or different risks when compared to the predicate device. The 2016 FDA guidance for human factors has not been updated, but FDA reviewers continue to issue deficiencies related to the objective evidence provided in a 510k for human factors validation.

The FDA must be consistent in the wording for “Hours for Receipt of Submission” because this affects submissions at the end of the fiscal year, but it also affects any submissions with a deadline for response to an RTA Hold, AI Response, and IDE submissions. The CDER and CBER divisions of the FDA address the need for defining the date of receipt in a guidance document specific to this topic, “ Providing Regulatory Submissions in Electronic Format–Receipt Date. ” Below is a screen capture copied from page 4 of the guidance.

Another element missing from this new guidance is a reference to human factors documentation. For any devices that have a user interface that is different from the predicate device, and for software devices, the FDA requires documentation of your human factors process to make sure that differences in the user interface do not result in new or different risks when compared to the predicate device. The 2016 FDA guidance for human factors has not been updated, but FDA reviewers continue to issue deficiencies related to the objective evidence provided in a 510k for human factors validation.
What are the new sections for a 510k submission?
In 2019, the FDA released a guidance document on the “ Format of Traditional and Abbreviated 510(k)s. ” That guidance outlines the 20 sections of a traditional 510k submission that have been used for decades. However, the new 510k electronic submission guidance has no numbering for the sections of the eSTAR template, and there are 22 sections instead of 20 sections. Several of the new sections are elements of the current FDA submission cover sheet (i.e., FDA Form 3514), and some sections exist in the 2019 guidance that were eliminated, such as: “Class III Summary and Certification.” Therefore, Medical Device Academy is recreating 100% of our 510k training webinars to explain how our 510k templates are used with the 510k eSTAR template and how to fill in the PDF form. To prevent confusion between the two formats, we are using letters for each section in the eSTAR template instead of numbers (i.e., A-V instead of 1-20). Table 1 from the new eSTAR guidance is reproduced below for your information.
Important information in the eSTAR guidance
In Table 1 above, there are 14 hyperlinks to various FDA guidance documents . These links are extremely helpful when you have questions about a specific question. Unfortunately, the 510k electronic submission guidance document will quickly become out-of-date as guidance documents are updated and made obsolete. In particular, one of the A-list final guidance documents that was planned for FY 2023 was the FDA cybersecurity guidance . The updated cybersecurity guidance was finally released last week.
9 thoughts on “510k Electronic Submission Guidance for FDA 510k Submissions”

Any updated info or guidance on where/how to document the human factors and usability documentation within the eSTAR?

That’s a great question. In the Performance testing section of the eSTAR template, the first question you are asked is: Was Bench Testing used in order to support this submission?
If you answer “yes” with the dropdown menu, then the eSTAR template will create a Bench Testing subsection. In that subsection there is a button for “Add Attachment.” That is where you attach the human factors and usability documentation within the eSTAR. The help button on the right provides you with a JavaScript Window that lists all of the different types of benchtop testing that you might be submitting. The last item in the list is:
“Usability/Human Factors: Studies specifically assessing the instructions and/or device design in terms of impact of human behaviour, abilities, and other characteristics on the ability of the device to perform as intended should be included here.”
The eSTAR version I’m quoting is nIVD v2.2. There is also a new draft guidance for the premarket submission documentation requirements for human factors. That gives you a good list of what the FDA wants in this section for attachments. When that guidance is finalized, I expect the FDA might expand the benchtop testing section to include a bunch of attachments much like you find in the software section. But that could be a year from now.
My detailed tutorial on how to add this information to the eSTAR will be covered in our March 16, 2023 live webinar. You can participate in that training if you purchase our 510(k) Course.
Thanks for the response- the IVD eSTAR is on version 1 which has none of these sections for non-clinical bench testing or human factors or usability. The first question in the Performance testing section is regarding Analytical Performance, and based on my previous answers for IVD software product, jumps to clinical studies, then performance testing summary.
You’re very welcome. I’m glad it was helpful to you.
So to follow up- any direction for the IVD eSTAR?
I don’t see anywhere in the eSTAR IVD v1.5 template that human factors is specifically mentioned either. I would expect it to belong in the performance testing section under the following question?
“Do you have other Clinical Supportive Data to include in this submission?”
This section allows you to attach multiple documents and explain the purpose of the documents in a text box above the attachments. This is what I would do for a device like a glucose test meter that requires human factors testing as part of the Special Controls.
I submitted this question to Lili Duan at the FDA, and she confirmed that this is correct…
“You are correct, the Human Factors / Usability information is attached to the Performance Testing à Clinical Studies à ‘Do you have other Clinical Supportive Data to include in this submission?’ question. Please note you can continue attaching additional bench tests, etc. by clicking on Add Attachment button and selecting files. However, please ensure that you provide a brief explanation of each attachment and what area it covers.”
Thanks Rob, for the update. So even though this is NON-clinical bench performance and usability (for SaMD, IVD) it should go in the Clinical Studies section?

Hi Rob, thank you so much for the videos, very informative. I quickly wanted to find out. What format will FDA except for 510K exempt as the eSTAR is mainly for premarket and De Novo.
There is no submission to the FDA for 510(k) exempt products at all. You may be required to have a Design History File (DHF) for Class 2 devices, but that DHF will be sampled during the routine inspections and there is no required format/content.
Leave a Comment Cancel Reply
Your email address will not be published. Required fields are marked *
Save my name, email, and website in this browser for the next time I comment.
This site uses Akismet to reduce spam. Learn how your comment data is processed .

Frequently Asked Questions
What information should be included in the 510(k) cover letter, 510(k) summary, and 510(k) statement.
The 510(k) cover letter should include information such as…
- Type of 510(k) submission
- Device type in plain terms
- 510(k) submitter name
- Contact information
- Recommended classification
- Device class
- Review panel
- FDA product code
- Preference for confidentiality
The 510(k) summary is similar, in that it is a brief summary of the device and the supporting information, while the 510(k) statement is a certification that the 510(k) holder will provide a copy of the submission to any FDA personnel within 30 days of a written request.
FDA’s 510(K) Submission Process
by OMC Medical | Jun 14, 2022 | FDA , USA

The FDA 510(K) Pre-market notification submission as per 21 CFR 807 Subpart E is to be adopted by manufacturers to receive FDA clearance to market medical devices or for commercial distribution in the U.S.
This review is done by FDA’s Centre for Devices and Radiological Health (CDRH). A 510(K) submission allows medical devices to be “ FDA Cleared ” and not FDA Approved.
The route to 510(K) must be carefully investigated by the manufacturer through a step-by-step process which allows determining if the regulatory pathway chosen for the Medical Device’s FDA access is in the right direction.
Step 1: Decision Criteria Checklist
The assessment checklist helps the manufacturer to arrive at a decision if they are eligible or fall under the rules to submit an FDA 510(K) application.
Step 2: Device Classification
The next step toward submission is to verify how the medical device is classified under the FDA classification regulations.
There are different generic types of devices identified by the FDA and placed under 3 categories of regulatory classes based on the risk posed by the medical device and the level of controls necessary for the safety and effectiveness of the device.
Class I Devices Low-Risk Devices – General Controls
- With Exemptions
- Without Exemptions
The above states that certain class I low-risk devices are exempted from the “General Controls”.
Class II Devices Moderately Risk Devices – General Controls and Special Controls
The above states that certain class II devices are exempted while others fall under the provisions of “General and Special Controls”.
Class III Devices High-Risk Devices – General Controls and Pre-market Approvals
- Pre-Market Approval
Step 3: Determine if your device is Substantially Equivalent to a Predicate Device
510(K) submission is applicable only for devices that can claim Substantial Equivalence (SE) to a predicate device. Below flowchart is an illustration that helps to clearly understand the decision route:
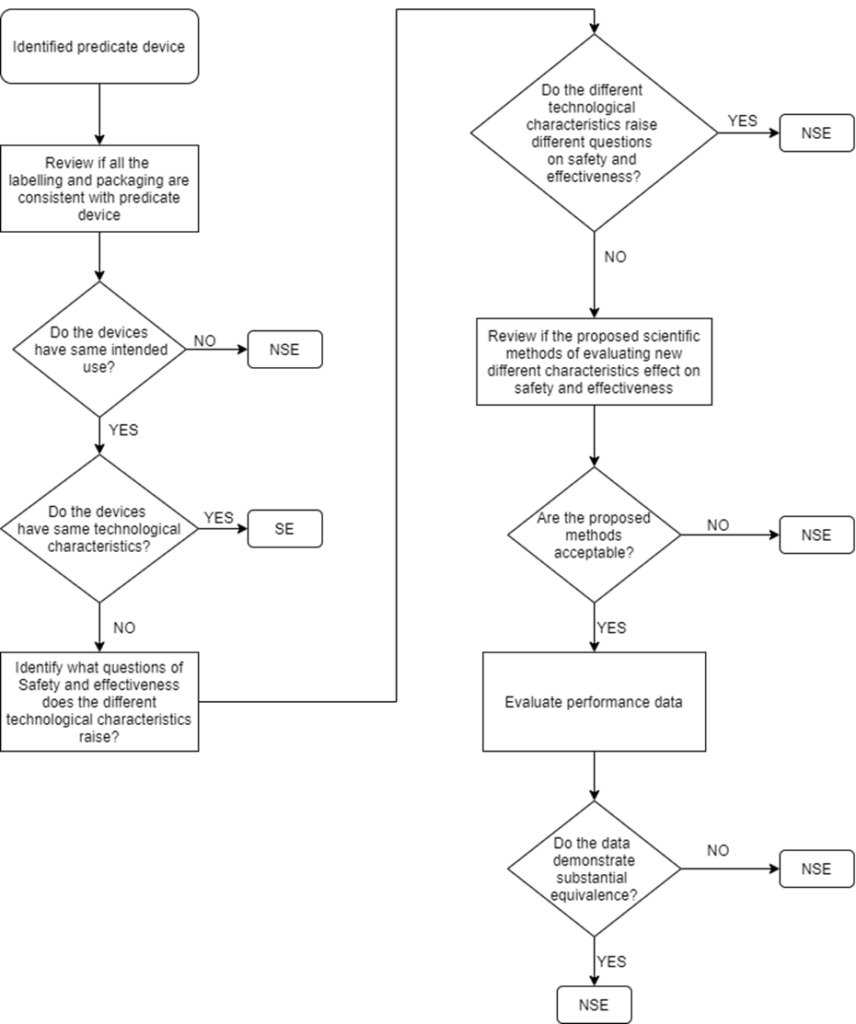
“SE” – Substantial Equivalent
“NSE” – Non-Substantial Equivalent
Multiple Predicate Devices
In certain cases, the manufacturer may identify more than one predicate device i.e., multiple predicates. In such cases, the primary predicate refers to the one that is most similar to the below factors:
- Intended Use
- Indications for use
- Technological characteristics
The manufacturer is recommended to identify the most appropriate primary predicate device with a well-supported decision document.
Supporting Documents to Claim Substantial Equivalence
The following are required by the manufacturer but not limited to, while demonstrating the most appropriate predicate device and that the new device to be submitted for 510(K) is a substantial equivalent to a predicate device.
- Indications for Use
- Technological Characteristics (similarities, differences and whether the differences pose different questions on safety and effectiveness)
- Performance data to support substantial equivalence (biocompatibility testing, Electrical safety and Electromagnetic Compatibility, Software verification and validation testing, mechanical testing, clinical study, animal study, if applicable)
- A declaration of conformity to recognised standards applicable to the medical device
Refer to the Section 807.92 content and format of a 510(K) summary.
Step 4: Determine the Type of 510(K) Submission
Within the 510(K) applications, there are 3 categories of submissions as discussed below
Step 5: The 510(K) Submission Process
Step 5.1 : form fda 3601 medical device user fee cover sheet.
Visit webpage User fee website to register and make payment.
Take a printed copy of this user fee cover sheet. This could be the first page of the 510(K).
Step 5.2 : FORM FDA 3514 CDRH PREMARKET REVIEW SUBMISSION COVER SHEET
Download form FDA 3514 pdf. This form captures detailed information required for the different types of submissions.
A cover letter and/or the FDA Form 3514 should follow the User fee cover sheet. If FDA Form 3514 is not affixed, then the cover letter should contain all the elements relevant to the submission contained in Form 3514. This will expedite the processing time.
Step 5.3: 510(K) Submission Acknowledgement receipt by FDA
If a valid eCopy and a proper user fee has been paid Acknowledgment Letter get received from DCC through email. If the proper fee and a valid eCopy are submitted by the holder, then the holder receives an acknowledgement letter from the DCC through an email.
The following are identified by the Acknowledgement Letter:
- Receipt’s date (the date that FDA received the 510(k) submission, an eCopy and the proper user fee payment);
- The receipt’s date (this is the day that 510(k) submission was received by FDA, valid eCopy, and proper user fee payment); and 510(k) number
Step 5.4: Document Contents in a 510(K) Submission
Below is the minimum list of contents necessary to be available in 510(K) submission documents.
- Cover Letter
- Table of Contents
- Indications for Use (FDA Form 3881)
- 510(K) Summary or Statement
- Truthful and accurate statement as required by 21 CFR 807.87(l)
- Class III Summary and Certification (to be submitted when claiming equivalence to a Class III device) As per 21 CFR 807.94
- Financial Certification & Disclosure Statement
- Declaration of Conformity and Summary Reports
- Proposed Labelling
- Sterilization and Shelf Life
- Biocompatibility
- Device Specifications
- Substantial Equivalence Comparison
- Electromagnetic compatibility and Electrical Safety
- Performance Data (Summary on Clinical and Non-Clinical data)
- Additional Requirements
Mode of Submission to FDA – E copies
In section 745A(b)(1) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) (21 U.S.C. 379k-1) FDA is amending its regulations on medical device submissions to remove requirements for paper and multiple copies and replace them with requirements for a single submission in electronic format.
Submissions in electronic format include eCopies, submissions created and submitted on CD, DVD, or flash drive and mailed to FDA, and eSubmissions, submission package produced by an electronic submission template.
Fees, Exemptions and Waivers
Under the user fee system, medical device companies pay fees to the FDA when they register their establishments and list their devices with the agency, whenever they submit an application or a notification to market a new medical device in the U.S. and for certain other types of submissions.
The MDUFA (Medical Device User Fee) website User Fee has information on the current fee charges applicable. Payment must be received and processed at the time or before the date the application is sent.
If the FDA receives an application without full payment of all required fees, the FDA will consider the application incomplete and will not begin its review.
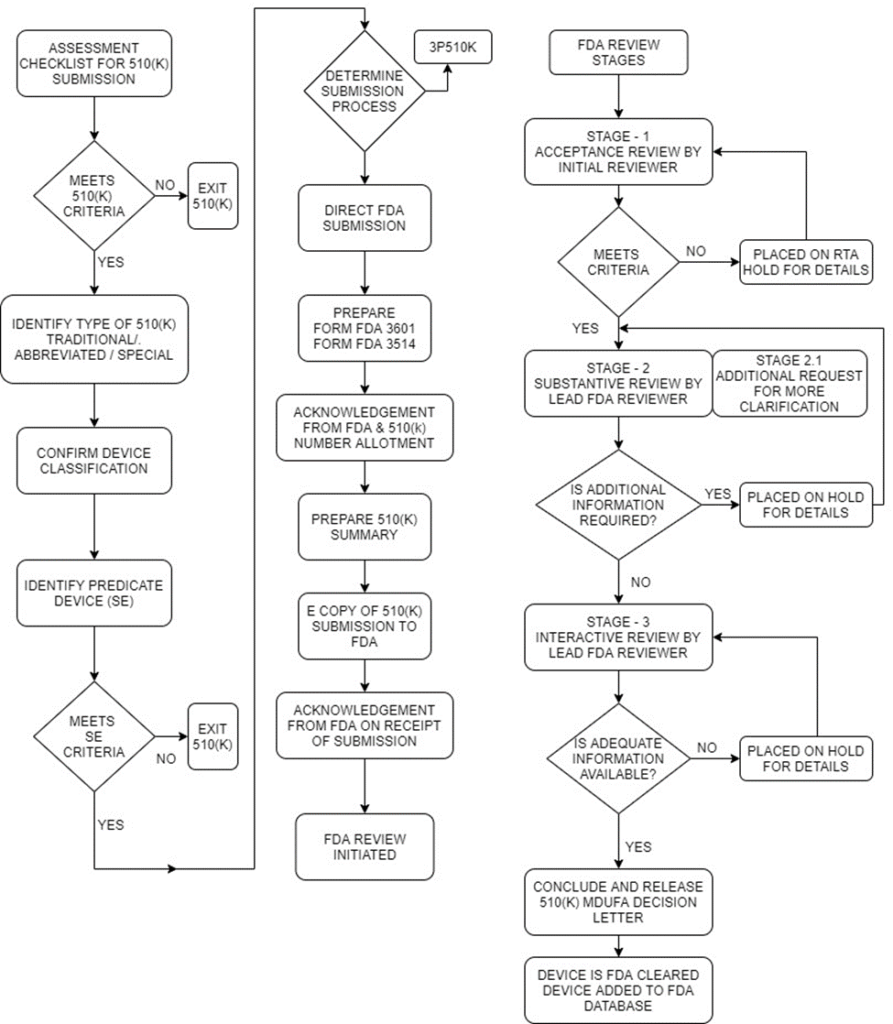
Review Stages
Acceptance review.
- This is based on the Refuse to Accept (RTA) policy by the FDA. It is a mechanism adopted by the FDA to provide a quick review of the 510(K) submission.
- This stage is only the initial (Acceptance review) stage where the FDA reviewer using separate checklists for each type of submission (traditional, abbreviated and special) reviews the submission and gives a declaration if the submission contents meet the minimum threshold requirements or is placed on RTA hold.
- The FDA reviewer evaluates the submission against specific acceptance criteria and informs the submitter within the above timeline on acceptance or indicate the missing element(s) in submission.
- In order to enhance the consistency of FDA’s acceptance decisions and to help submitters better understand the types of information FDA needs to conduct a substantive review, this guidance, includes the checklists to clarify the necessary elements and contents of a complete 510(k) submission.
Only if this stage is cleared, the submission gets qualified for the actual Substantive review stage. The reviewer conducting substantive review is the actual Lead reviewer.
Substantive Review
During Substantive Review, the Lead Reviewer conducts a comprehensive review of the 510(k) submission and communicates with the submitter through a Substantive Interaction, which should occur within 60 calendar days of receipt of the 510(k) submission.
Substantive Interaction communication is typically:
- an email stating that FDA will proceed to resolve any outstanding deficiencies via Interactive Review; or
- an Additional Information (AI) request which places the submission on hold.
Additional Information (AI) request
If the lead reviewer sends an AI request, then it means the submission is placed on hold. The submitter has 180 calendar days to address the queries from the date of additional information request by FDA reviewer.
If the queries are not addressed by the applicant within this time span, then 510(K) submission is deleted from the FDA database and the applicant will need to submit a new 510(K) to pursue the FDA market clearance process.
The submitter must submit the response, with a valid eCopy, to the DCC. The response should:
- include the submitter’s name;
- list the 510(k) number;
- identify the submission as Additional Information (AI) to the 510(k);
- list the date of FDA’s request for additional information; and
- provide the requested information in an organized manner.
Interactive Review
If the Lead Reviewer chooses to continue with an Interactive Review, this means the Lead Reviewer has determined that any outstanding deficiencies may be adequately addressed within the timeframe set by the Medical Device User Fee Amendment of 2012 (MDUFA III) performance goal for a 510(k) (90 FDA days) and that the submission will not be placed on hold.
The Lead Reviewer communicates with the submitter during the Interactive Review using tools such as:
- Telephone Call
During Interactive Review, the Lead Reviewer may request additional information from the submitter, who may either send the information to the Lead Reviewer directly or to the DCC (Document control centre).
Note: During Interactive Review, any information submitted to the DCC must include a valid eCopy.
Timeline – An overview of 510(K) Submission
FDA receives the 510(K)-application submission
FDA sends the acknowledgement letter (or) FDA sends HOLD letter (in case of issues)
FDA conducts Acceptance Review and informs the applicant if the application is eligible for substantive review (or)
Places it under RTA Hold
FDA conducts Substantiative Review and communicates on the next move towards Interactive Review
FDA sends Final MDUFA Decision Letter
Step 6: Final 510(K) Decision Letter
MDUFA Decisions for 510(k) submissions include findings of substantially equivalent (SE) or not substantially equivalent (NSE).
When a decision is made, FDA will issue the decision letter to the submitter by email to the email address provided in the 510(k) cover letter.
- A 510(k) that receives an SE decision is considered “cleared.”
- FDA adds the cleared 510(k) to the 510(k) database, which is updated weekly.
- The IFU and the summary will be sent as attachments to the SE letter. The IFU will not be signed since it is considered an attachment to the SE letter. Therefore, the signature on the SE letter will apply to both the letter and the IFU.
If FDA does not reach an MDUFA decision within 100 FDA days (i.e., 10 days after the MDUFA goal), FDA will issue a Missed MDUFA Communication, which is written feedback to the submitter to be discussed in a meeting or teleconference, including the major outstanding review topic areas or other reasons that are preventing FDA from reaching a final decision, with an estimated date of completion.
510(K) decision-making flow chart
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Submit a Comment Cancel reply
Your email address will not be published. Required fields are marked *
Save my name, email, and website in this browser for the next time I comment.
Recent Posts
- Bio Korea 2024 – The Future of Biotechnology Innovation and Global Collaboration
- Directive (EU) 2019/904 on the reduction of the Impact of certain Plastic Products on the Environment
- Iraq Medical Device Registration: KIMADIA Guidelines and Requirements
- License Transfer for Medical Device to New Sponsors from Existing License Holders
- License Transfer for Medical Device to new Local AR – New Zealand
Recent Comments

- Search forums
- National and International Business System Standards
- Food and Drug (Pharmaceuticals) related Regulations
- US Food and Drug Administration (FDA)
FDA 510(k) Cover Letter Contents
- Thread starter ritammy
- Start date Feb 1, 2013
- Feb 1, 2013
I work for a ISO 13485 certified medical device company. We are designing our first product that requires a 510(k). Wondering if anyone has a sample of what the FDA is looking for with a Cover Letter. This will be the first 510(k) I am compiling. I have guides to what to include in all the other areas but nothing can be found to show what a 510(k) cover letter should look like and what data should be included. Thanks!
Waterman956
Here is an example, the FDA address has change so make sure and change it
Attachments
- 510k Cover Letter.docx 15.8 KB · Views: 1,719
Thanks so very much. This is very helpful. Have a great weekend!
Similar threads
- BuckeyeBurro
- May 5, 2024
- Jan 18, 2024
- US Medical Device Regulations
- Sep 20, 2023
- Oct 19, 2023
- Medical Device and FDA Regulations and Standards News
- franklymissshankly
- Jun 8, 2022
- This site uses cookies to help personalise content, tailor your experience and to keep you logged in if you register. By continuing to use this site, you are consenting to the use of cookies. Accept Learn more…
- Top Courses
- Online Degrees
- Find your New Career
- Join for Free
How to Write a Cover Letter When You’re Changing Careers (Sample + Tips)
As a career changer, you need to help recruiters understand why you’re moving away from your former line of work and what you want to achieve in your new career path..
![[Featured Image] A man in a blue button-up is sitting down in a conference room holding pieces of paper.](https://d3njjcbhbojbot.cloudfront.net/api/utilities/v1/imageproxy/https://images.ctfassets.net/wp1lcwdav1p1/28u80RCd3SjJJ03qjwbPZJ/95337dc542ebaf56e3e04ba4835c2bab/9T9Z7AiJ.jpeg?w=1500&h=680&q=60&fit=fill&f=faces&fm=jpg&fl=progressive&auto=format%2Ccompress&dpr=1&w=1000 "cover letter 510k")
You will inevitably change jobs throughout your career as you seek more responsibility, growth, or even a higher salary. According to the US Bureau of Labor Statistics, the average employee stays at each job for around four years [ 1 ]. However, for career changers—or those interested in exploring an entirely new path or industry—making that switch can sometimes involve unique challenges.
Even so, making a career change has become an increasingly popular move. More than half of workers in the United States anticipated looking for a new opportunity in 2022 [ 2 ]. Changing careers can allow you to find more meaningful work, better align your career path with your larger goals, and move into a more energizing role.
When you draft your cover letter to apply for a job in a new line of work, you must take time to explain your larger objectives. In this article, we’ll review specific information you can feature in your cover letter to help recruiters understand your goals and reasons for changing careers.
Learn more: How to Plan for a Career Change: Step-by-Step Guide
How to write a career change cover letter
A cover letter is a chance to expand upon the bullet points outlined in your resume . It’s a space where you can explain your interest in the role and company, highlight your experience and skills, and sell a recruiter on the overall fit you’d make.
But a career changer needs to do all of that and more. You also need to help recruiters and hiring managers understand why you’re moving away from your former line of work, what you want to achieve in your new career path, and any transferable skills that will help make your transition smooth.
Let’s review four key pieces of information you can weave into your career change cover letter.
1. Clarify your career change context
Explaining why you’re interested in changing careers and how the role you’re applying to fits within your larger career aspirations can preemptively contextualize your story. Plan to include a career change objective somewhere in your cover letter, much like you would a resume objective to provide a summary of a person’s experience and goals. Don’t be afraid to build a sense of personality so recruiters can better connect you with your objective.
What this looks like: I’ve spent the last six years translating complex topics for various users as a technical writer. But in that time, I’ve realized that what drives me is the user’s experience. It’s the lightbulb moment behind my career change to UX design . I believe I’ll make a strong addition to your team because my work has largely put the user front and center, and now I’m interested in focusing on a different facet of that goal.
2. Specify the value of your certificates, courses, or trainings
It costs over $4,000 to hire an employee, according to the Society for Human Resources Management [ 3 ]. That’s all the more reason why recruiters and hiring managers want to find the right candidate. It can be costly otherwise. Help explain what you’ve done to prepare for your career change by highlighting any professional certificates or trainings you’ve completed to prepare you for your new line of work.
What this looks like: In order to familiarize myself with the tools and processes used in data analysis, I completed the Google Data Analytics Professional Certificate , which taught me SQL and R, and trained me to clean and visualize data. Thanks to this preparation, I feel confident that I will make a strong addition to your team from the very start.
3. Bring attention to your transferable skills
Transferable skills are “portable,” in that you take them from job to job. They include problem-solving, critical thinking, attention to detail, and more. Show recruiters that you have important skills to help you do the job so they can understand the unique value you’d bring to their company.
It can also help to find out the key technical skills the job requires and spend time learning what you can, especially when it comes to important software or tools.
What this looks like: As a software developer, I regularly relied on my problem-solving skills to think through complex issues. I’ll bring that same skill, as well as my attention to detail, listening, and decision-making, to ABC High School as the new algebra teacher.
4. Highlight your past achievements
Any time you can highlight what you’ve managed to accomplish in your past roles, you help a recruiter see your potential in a new role. Where possible, summarize any moments that showcase your strengths and illustrate your work ethic or character.
What this looks like: I pride myself on being a team player and a problem-solver. As a social media manager at Company X, I identified a better program to help my team schedule content. Using that tool improved my team’s efficacy, leading to our most successful quarter.
Why is a cover letter important when changing careers?
The idea of a career path can sometimes be rigid, suggesting that people only follow one specific track. Although that perspective is starting to shift, it’s still prevalent. You can help recruiters and hiring managers understand more about your interest in a role by explaining why you’re changing careers and what you’ve done to streamline your transition.
It helps to align your cover letter with a resume objective, which can be especially useful for career changers. An objective on your resume is a place where you can contextualize your larger career aims, quickly summarizing what you’re hoping to achieve in your next role. Repeat that same information in your cover letter and expand on it slightly to give your application materials more cohesiveness.
Read more: How to Use Resume Sections to Shape Your Professional Story

Build job-ready skills with a Coursera Plus subscription
- Get access to 7,000+ learning programs from world-class universities and companies, including Google, Yale, Salesforce, and more
- Try different courses and find your best fit at no additional cost
- Earn certificates for learning programs you complete
- A subscription price of $59/month, cancel anytime
Career change cover letter sample
It's common practice nowadays to submit your cover letter digitally. In that case, include some of your contact information in the top left corner so recruiters can easily see how to get in touch.
Thomas Bennett
Nashville, TN
(555) 555-1234
Dear Ms. Tufte,
I’m writing to apply for the project manager role at Company X. I initially began my career as a marketing coordinator and eventually moved into email marketing , where I was responsible for strategizing and developing new campaigns. But in that time, I realized how much I thrived when managing our quarterly campaigns from start to finish. That’s why I’m interested in segueing into project management.
Knowing that, despite my experience, I still needed to learn more specifically about project management, I completed the Google Project Management Professional Certificate . Over six months, I’ve learned Agile project management as well as how to create product documentation, among other key skills. I believe this training, along with my previous experience, will help me transition to a project management role at Company X and make a big impact.
I’m an organized problem-solver with a sharp eye for detail, all important project management skills. I believe my previous work in email marketing provided hands-on training in managing projects, albeit without the official title. I identified new tools to help my team create more effective quarterly campaigns. As a result, we increased our click-through rate (one of our key metrics) to 1.87 percent, bringing it closer to the industry standard—an immense achievement.
I’m proud of the foundation I gained through marketing, but in realizing where my true passion lies, I’m keen to transition into a project management role with more growth opportunities. I appreciate your consideration.
Tips for strengthening your cover letter
Much like you would for a standard cover letter, you can strengthen your cover letter as a career changer using the following tips:
Tailor your letter for each role.
You should tailor your resume for each role you apply to, and the same goes for your cover letter. Research the company, find out about aspects of their work that interest you, and insert those details into your cover letter. You should also tailor your experience and skills, highlighting each job's most relevant skills and accomplishments.
Get specific.
Your cover letter should expand upon your resume rather than repeating the same information. One way to do this is by giving details about your past achievements. When possible, quantify your impact with numbers and explain how these accomplishments make you uniquely qualified for this new role.
Use action words.
Build action words into your resume and your cover letter. Rather than more staid words that don’t capture your unique story or responsibilities, action verbs can liven up your cover letter and make it more enticing to read. Find verbs that succinctly and accurately depict your previous experience.
Start advancing your skills today
Brush up on your cover letter writing skills by taking the University of Maryland’s free course, Writing Winning Resumes and Cover Letters . Or develop important skills for an in-demand career with a Professional Certificate from industry leaders like Google, Meta, and IBM. Most certificate programs take less than seven months to complete, and you can start for free with a seven-day, all-access trial.
Article sources
US Bureau of Labor Statistics. “ Employee Tenure in 2020 , https://www.bls.gov/news.release/pdf/tenure.pdf.” Accessed April 26, 2024.
CNBC. “ The Great Resignation is Likely to Continue , https://www.cnbc.com/2021/08/25/great-resignation-55-percent-are-looking-to-change-jobs-over-the-next-year-.html.” Accessed April 26, 2024.
ADP. “ Calculating the True Cost to Hire Employees , https://www.adp.com/spark/articles/2019/07/calculating-the-true-cost-to-hire-employees.aspx.” Accessed April 26, 2024.
Keep reading
Coursera staff.
Editorial Team
Coursera’s editorial team is comprised of highly experienced professional editors, writers, and fact...
This content has been made available for informational purposes only. Learners are advised to conduct additional research to ensure that courses and other credentials pursued meet their personal, professional, and financial goals.
The Federal Register
The daily journal of the united states government, request access.
Due to aggressive automated scraping of FederalRegister.gov and eCFR.gov, programmatic access to these sites is limited to access to our extensive developer APIs.
If you are human user receiving this message, we can add your IP address to a set of IPs that can access FederalRegister.gov & eCFR.gov; complete the CAPTCHA (bot test) below and click "Request Access". This process will be necessary for each IP address you wish to access the site from, requests are valid for approximately one quarter (three months) after which the process may need to be repeated.
An official website of the United States government.
If you want to request a wider IP range, first request access for your current IP, and then use the "Site Feedback" button found in the lower left-hand side to make the request.
- Skip to main content
- Skip to FDA Search
- Skip to in this section menu
- Skip to footer links

The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
U.S. Food and Drug Administration
- Search
- Menu
- Medical Devices
- Products and Medical Procedures
- Device Approvals and Clearances
- 510(k) Clearances
Downloadable 510(k) Files
You can download any of the following zipped files, each of which contains information about the releasable 510(k)s for the time frame indicated. Each record in the file is 272 characters in length. These files are replaced monthly usually on the 5th of each month. In addition there is a file description and an explanation of some of the codes used in the file. You can also download or search the Product Code Classification Database.
Most current month available: PMNLSTMN.ZIP
1996-current: PMN96CUR.ZIP
1991-1995: PMN9195.ZIP
1986-1990: PMN8690.ZIP
1981-1985: PMN8185.ZIP
1976-1980: PMN7680.ZIP
Additional Information
- File Layout for Releasable 510(k)s
IMAGES
VIDEO
COMMENTS
The statement may be included in the 510(k) Cover Letter or may be on a separate page identified in the table of contents. If the CDRH Premarket Review Submission Cover Sheet is used, the ...
Section 3.0 - 510(k) Cover Letter. Section 4.0 - Indications for Use Statement. Section 5.0 - 510(k) Summary. Section 3.0 is exactly what it sounds like: a cover letter with some basic administration information, the basis for the submission, and a table supplying information on the design and use of the device. It should be kept fairly ...
This document supersedes "Format for Traditional and Abbreviated 510(k)s - Guidance for Industry and FDA Staff" issued November 17, 2005. For questions about this document, contact ORP: Office of Regulatory Programs, DRP1: Division of Submission Support, Premarket Notification and Classification Team at. [email protected] or 301-796 ...
510k Cover Letter Webinar ($29) 510k Cover Letter Webinar. In this 20 minute webinar you will learn how to prepare a 510k Cover Letter and complete FDA Form 3514-including identification of recognized standards. You will also receive copies of the templates for both documents--updated for 2020. Price: $29.00.
Cover letter; Statement of indications for use; 510(k) summary OR statement . Your cover letter is a broad summary of your 510(k) submission. Your indications of use statement MUST match the indications for use of your predicate device. It's made public after 30 days - so don't reveal too much if your device is designed to fill a market niche!
510(k) Cover Letter - Identify 510(k) holder - Type submission - Associated submissions - Basis for submission • Executive Summary - concise description of the device, including the indications for use and technology; - device comparison table; and - concise summary for any performance testing in the submission. What Should Be ...
Prepare "abbreviated 510(k)" cover sheet. ... Typically, you will write the cover letter last, so as to provide a good, high-level summary. Your cover letter should include the following: Type of 510(k) submission, Abbreviated or Traditional. Your device type in plain terms, i.e., by its common name. 510(k) submitter.
Identification of key information that may be useful to FDA in the initial processing and review of the 510(k) submission, including content from current Form FDA 3514, Section A. B: Cover Letter / Letters of Reference: Attach a cover letter and any documents that refer to other submissions. C: Submitter Information
Cover Letter Table of Contents 510(k) Acceptance Checklist Indications for Use Statement 510(k) Summary Declaration of Conformity Device Description: Narrative and Physical Substantial Equivalence Discussion Proposed Labeling Performance Testing Regulatory Best Practices Guide Types of 510(k) Submissions Traditional 510(k) - the most
There is no 510(k) form; however, 21 CFR 807 Subpart E describes requirements for a 510(k) submission. Before marketing a device, each submitter must receive an order, in the form of a letter ...
The 510 (k) cover letter should include information such as…. The 510 (k) summary is similar, in that it is a brief summary of the device and the supporting information, while the 510 (k) statement is a certification that the 510 (k) holder will provide a copy of the submission to any FDA personnel within 30 days of a written request.
The receipt's date (this is the day that 510(k) submission was received by FDA, valid eCopy, and proper user fee payment); and 510(k) number; Step 5.4: Document Contents in a 510(K) Submission. Below is the minimum list of contents necessary to be available in 510(K) submission documents. Cover Letter; Table of Contents
510(k) Number (if known) K180421 Device Name Natus NeuroWorks Indications for Use (Describe) The NeuroWorks is EEG software that displays physiological signals. The intended user of this product is a qualified medical practitioner trained in Electroencephalography. This device is intended to be used by qualified medical ... 510(k) PMN Cover ...
Feb 1, 2013. #1. I work for a ISO 13485 certified medical device company. We are designing our first product that requires a 510 (k). Wondering if anyone has a sample of what the FDA is looking for with a Cover Letter. This will be the first 510 (k) I am compiling. I have guides to what to include in all the other areas but nothing can be found ...
510(k) No: _____ System: EPIQ 5 and EPIQ 7 Ultrasound Systems Intended Use: Diagnostic ultrasound imaging or fluid flow analysis of the human body as follows: Clinical Application Mode of Operation (*includes simultaneous B-mode) General (Track I only) Specific (Tracks I & III) B M PWD CWD Color Doppler* Combined (Spec.) Other (Spec.)
Let's review four key pieces of information you can weave into your career change cover letter. 1. Clarify your career change context. Explaining why you're interested in changing careers and how the role you're applying to fits within your larger career aspirations can preemptively contextualize your story.
Exempt Device Review Form (PDF - 16KB) 510 (k) Cover Sheet Memorandum (PDF - 41KB) 510 (k) "Substantial Equivalence" Decision Making Process (PDF - 844KB) Indications for Use (PDF - 1.7MB ...
3. 5 10(k) Notification Cover Letter 3.1. Administrative InformationFDCRDM Type: Traditional 5 10(k) submission JN 20 Device type: accelerator, linear, medical 510(k) submi~tter: Naslund Medical AB Contact persons: Tomas Naslund No confidentiality needed concerning the existence of this premarket notification Regulation Number: 892.5050 Class: 2
This PDF is the current document as it appeared on Public Inspection on 04/29/2024 at 8:45 am. It was viewed 5778 times while on Public Inspection. If you are using public inspection listings for legal research, you should verify the contents of the documents against a final, official edition of the Federal Register.
The .gov means it's official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.